
On distingue les pieds bots idiopathiques
et les pieds bots secondaires
Qu’est-ce qu’un pied bot idiopathique ?
Le terme de pied bot idiopathique signifie qu’il n’existe pas d’autre maladie
pouvant participer à la déformation du pied.
Embryologie
Le développement du pied pendant la croissance intra-utérine (morphogenèse) est très précoce : pendant le 2ème mois de vie.
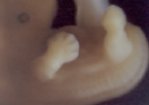

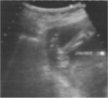
Diagnostic ante-natal
Actuellement 46 % des pieds bots varus équin sont diagnostiqués en échographie ante-natale
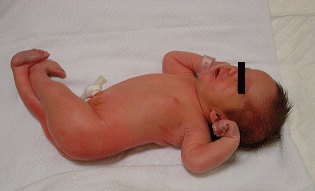
Le pied bot, appelé aussi pied bot varus équin, est une anomalie de développement du pied survenant pendant la période fœtale.
Il s’agit d’une déformation congénitale (c’est à dire existant dès la naissance).
Fréquence
Cette anomalie touche 1 à 2 enfant(s) pour 1000 naissances.
Sexe
Les garçons sont plus touchés que les filles (70% des cas).

Côté
Les deux pieds sont touchés dans la moitié des cas.
Les os du pied, leurs articulations et les parties molles (tendons, muscles, peau, capsules des articulations) sont déformés dans plusieurs plans de l’espace.
Ces déformations sont visibles dès la naissance.
Le pied et la cheville manquent de souplesse et semblent enraidis en mauvaise position.
Aspects génétiques du pied bot varus équin
Dans certaines familles et populations, la survenue de pied bot varus équin idiopathique est plus fréquente.
La recherche de facteurs génétiques (chromosomiques) favorisant a déjà fait l’objet de plusieurs publications.
Voir la page Génétique du pied bot (en cours de réalisation)
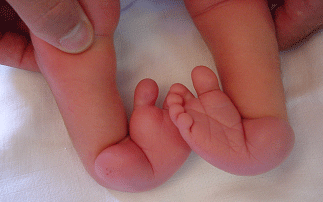
Pour faire le diagnostic de pied bot à la naissance, il faut constater une déformation et une raideur du pied dans plusieurs plan de l’espace :
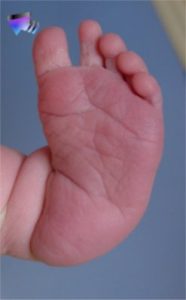 adduction de l’avant-pied
adduction de l’avant-pied
en regardant le pied par le dessous, on voit que la partie avant du pied (avec les orteils) tourne à l’intérieur
(Glossaire)
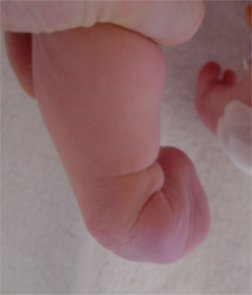 varus de l’arrière -pied
varus de l’arrière -pied
en regardant le pied par derrière, on voit que le talon bascule à l’intérieur
(Glossaire)
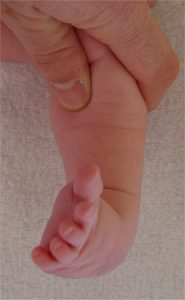 supination du pied
supination du pied
en regardant le pied par devant, on voit que la plante du pied se tourne vers le haut
(Glossaire)
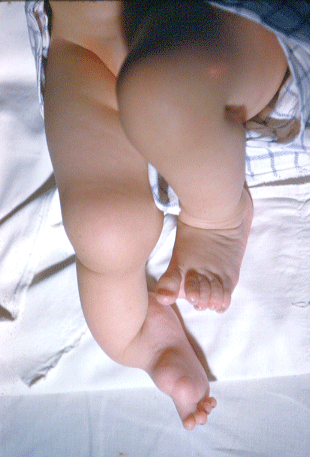
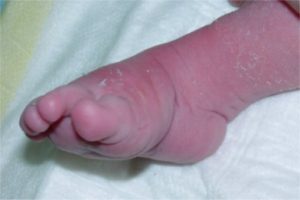 équin du pied et de la cheville
équin du pied et de la cheville
en regardant le pied de profil, on voit que la pointe du pied est dirigée vers le bas
(Glossaire)
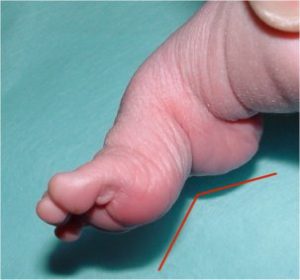 cavus du pied
cavus du pied
le cavus est l’aspect de cassure du pied, nettement visible au niveau du milieu du pied, sur son bord interne
(Glossaire)
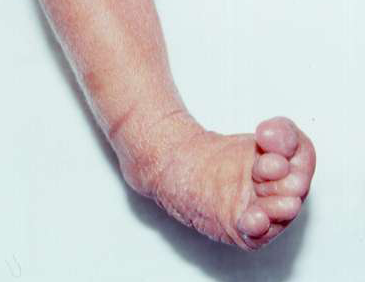
Le pied varus
Il s’agit d’une malposition du pied qui ressemble beaucoup au pied bot varus équin, à la différence que cette mauvaise position est réductible lors de la mobilisation passive du pied.
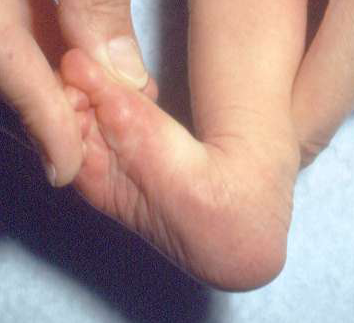
Le métatarsus adductus
Il s’agit d’une malposition du pied caractérisée par une bascule en dedans de la partie avant du pied par rapport à la partie arrière.
C’est aussi une des composantes du pied bot varus équin.
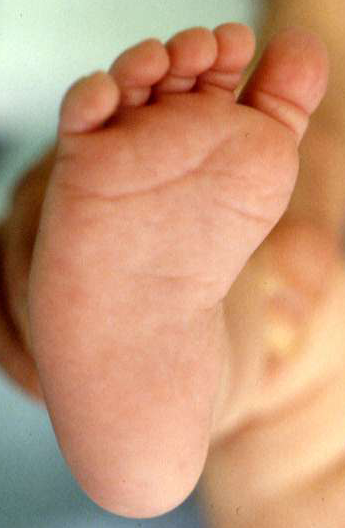
Le pied convexe
Il s’agit d’une déformation du pied plus rare que le pied bot varus équin, caractérisée par une plante de pied arrondie (convexe vers le bas), et des anomalies radiographiques.
Le traitement est difficile et souvent chirurgical.
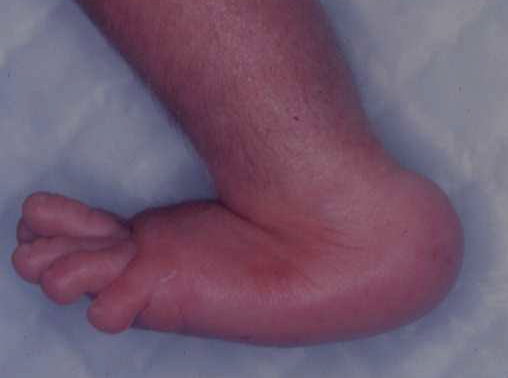
Le pied talus direct
Il s’agit d’une malposition du pied caractérisée par une flexion dorsale excessive faisant que le dos du pied vient toucher la face antérieure de la jambe.
Le pronostic sous traitement (rééducation et attelles) est excellent.
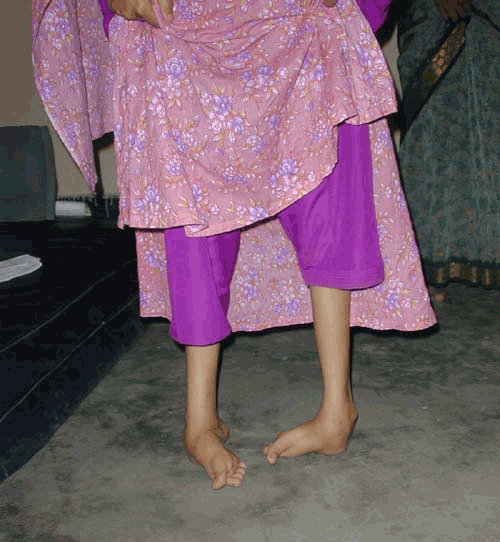
Evolution naturelle du pied bot (sans traitement)
Le pied bot idiopathique n’a pas de tendance naturelle à la correction spontanée.
Avec le temps, les déformations se figent et les articulations deviennent de plus en plus rigides.
La marche est possible mais se fait sur le bord externe des pieds, voire le dos des pieds, ce qui est source de douleurs.
A ce stade d’évolution, même la chirurgie traditionnelle du pied bot n’est pas possible et il faut corriger les pieds par des appareillages extérieurs (fixateurs externes)
Qu’est-ce qu’un pied bot secondaire ?
Un pied bot secondaire (ou syndromique) apparaît dans un contexte de maladie plus générale du corps :
maladie neurologique, maladie touchant la souplesse de tous les tissus.
Les pied bots secondaires sont souvent très difficiles à soigner.
Environ 10% des pieds bots varus équins sont non idiopathiques ou syndromiques.
Voici les syndromes où l’on peut retrouver un pied bot varus équin :
Dans un premier tableau, les syndromes où le pied bot est retrouvé de façon fréquente.
Dans un second tableau, les syndromes où le pied bot est retrouvé de façon occasionnelle.
Agénesie sacrée et Spina Bifida
Le Spina Bifida est le plus fréquent des défauts du tube neural. Le tube neural est la structure embryonnaire qui se développe entre le cerveau et la moelle épinière.
Le Spina Bifida concerne les vertèbres et parfois aussi la moelle épinière.
Le Spina Bifida « Aperta » désigne une ouverture dorsale (postérieure) des vertèbres associée à une atteinte plus ou moins prononcée de la moelle épinière. Il peut se former une poche au niveau cutané, contenant les méninges seules (méningocèle) ou associée à la moelle (myélo-méningocèle).
Il se situe le plus souvent dans la partie lombaire ou sacrée du rachis, sur 2 à 3 vertèbres, parfois plus. Les conséquences de ce trouble du développement vertébro-médullaire sont une paraplégie (paralysie des membres inférieurs), une hydrocéphalie et une malformation d’Arnold Chiari (conséquence de la moelle attachée pendant la vie intra-utérine), une incontinence urinaire et anorectale.
Ces troubles sont d’intensité très variable selon le niveau de la lésion et son étendue.
Cette malformation a une fréquence de 0,5 pour 1000 en France. En Europe la prévalence moyenne enregistrée pour le Spina Bifida est 5 sur 10 000 naissances.
En période néonatale, la neurochirurgie est palliative : elle vise à refermer le plan cutané et une dérivation ventriculopéritonéale est posée s’il existe une hydrocéphalie. Ultérieurement de nombreuses prises en charge sont nécessaires.
L’étiologie est multifactorielle. Le risque de récurrence lors d’une prochaine grossesse peut être diminué par la prise d’acide folique (vitamine B9) avant et en tout début de grossesse. Un diagnostic prénatal peut être réalisé par échographie.
En savoir plus sur le spina bifida
Anomalie du chromosome 12
Aplasie du tibia avec ectrodactylie
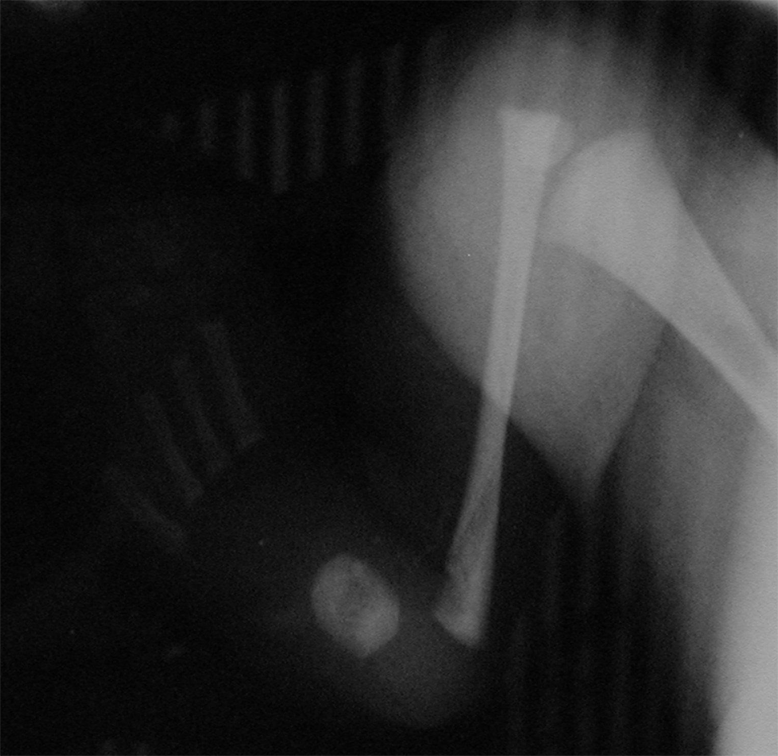
Arthrogrypose (Distale ou Arthrogrypose multiple congénitale)
L’arthrogrypose est une affection non héréditaire congénitale (existante à la naissance). Elle se caractérise par des malpositions des membres prédominantes aux extrémités et souvent symétriques. Dans 45% des cas, elle touche les quatre membres, dans 45% les membres inférieurs, et les membres supérieurs seuls dans 10%. L’aspect du visage de l’enfant est souvent caractéristique et l’intelligence habituellement normale.
Le tableau évocateur d’arthrogrypose :
– mobilité active et passive des articulations est réduite
– force musculaire est diminuée
– pas de trouble de la sensibilité
– plis de flexion articulaire sont absents, en particulier aux genoux, au creux inguinal, aux coudes et aux doigts
– la peau est tendue, luisante
– abondance de la graisse sous cutanée qui donne un aspect tubulaire aux membres
– fossettes à proximité des articulations, à la face antérieure des genoux, à la face postérieure des coudes mais aussi sur les épaules et sur les poignets
– visage est arrondi, souvent sans expression
– taches angiomateuses très caractéristiques à la pointe ou à la racine du nez qui ont tendance à s’estomper à mesure que l’enfant avance en âge.
En savoir plus sur l’arthrogrypose
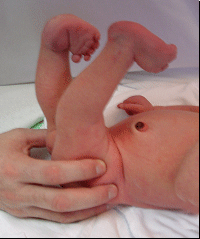
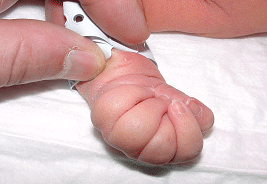
Atélostéogenèse
Atélostéogenèse de type 1
L’atélostéogenèse de type 1 (AO1, formation incomplète des os) est une dysplasie squelettique du nouveau-né à mortalité péri-natale caractérisée par des membres courts, un faciès typique et une radiologie signant le diagnostic. Il s’agit d’une maladie très rare. L’ AO1 et la dysplasie en boomerang résultent de mutations hétérozygotes dans le gène codant pour la filamine B (FLNB). Tous les cas de cette maladie à transmission autosomique dominante ont été sporadiques car résultant vraisemblablement de mutations de novo dans FLNB. L’échographie gestationnelle peut déceler une dysplasie osseuse. Le diagnostic précis peut être établi après la naissance par radiographie osseuse et par examen histologique du tissu ostéo-cartilagineux. Les patients naissent mort-nés ou meurent rapidement après la naissance. Il existe un continuum entre l’atélostéogenèse de type 1 et la dysplasie en boomerang, et les deux intègrent un même groupe nosologique. *Auteurs : Pr. D. Sillence et Pr. K. Kozlowski (novembre 2004).
Atélostéogenèse de type 2
L’atélostéogenèse de type II est une dysplasie périnatale caractérisée par un raccourcissement des membres, un syndrome dysmorphique et un aspect radiologique permettant d’établir le diagnostic. En effet, le diagnostic peut être fait sur la base d’un examen radiologique de la totalité du squelette. Les sujets atteints sont mort-nés ou décèdent peu de temps après la naissance. Cette maladie est rarement rapportée. Elle se transmet de manière autosomique récessive. L’atélostéogenèse de type II résulte des mutations du gène codant pour le transporteur du sulfate de la dysplasie diastrophique (DTDST) et est associée, d’un point de vue physiopathologique, au phénotype modéré de la dysplasie diastrophique et au phénotype sévère de l’achondrogenèse IB. Le diagnostic prénatal par étude moléculaire de l’ADN par biopsie du trophoblaste au cours du premier trimestre de la grossesse est possible lorsque les mutations de DTDST sont identifiées. *Auteurs : Prof. D. Sillence et Prof. K. Kozlowski (novembre 2004) Traduction Orphanet*.
Atélostéogenèse de type 3
L’atélostéogenèse de type III (AOIII, ossification incomplète) est une dysplasie caractérisée par un raccourcissement des membres, une dysmorphie faciale et une radiologie caractéristique. L’insuffisance respiratoire récidivante (trachéo-broncho-malacie) et/ou les infections sont les causes du décès précoce lié à cette maladie. Il s’agit d’un syndrome très rarement rapporté. L’atélostéogenèse de type III résulte de mutations hétérozygotes du gène codant pour la filamine B (FLNB). Tous les cas de ce syndrome autosomique dominant sont sporadiques. Le diagnostic peut être établi sur la base d’un examen radiologique de la totalité du squelette. Le diagnostic prénatal peut être fait par échographie gestationnelle. *Auteurs : Prof. D. Sillence et Prof. K. Kozlowski (novembre 2004) Traduction Orphanet*.
Dysostose Huméo-spinale
Dysplasie campomélique
Le syndrome campomélique est une atteinte très rare, caractérisée par l’association variable d’anomalies squelettiques (incurvation et gracilité des os longs , anomalies du bassin et du thorax, présence de 11 paires de côtes) à des anomalies extrasquelettiques (dysmorphie faciale, fente palatine, ambiguïté ou réversion sexuelle chez 2/3 des garçons, malformations cérébrales, cardiaques et rénales).
Sa prévalence à la naissance est de 1/300 000 environ.
Le syndrome est associé à des anomalies du gène SOX9, localisé en 17q24, soit lors de réarrangements chromosomiques impliquant son locus (au milieu ou à distance du gène), soit par mutation hétérozygote de ce gène survenue de novo. Le diagnostic est généralement évoqué lors de l’échographie morphologique du 2ème trimestre de la grossesse devant l’association d’un retard de croissance, d’anomalies osseuses avec incurvation des membres et éventuellement d’un pseudohermaphrodisme masculin. Une étude génétique prénatale peut être réalisée par amniocentèse ou sur un prélèvement de villosité choriale en cas de suspicion diagnostic ou dans les cas familiaux où une anomalie de cette région chromosomique a déjà été identifiée.
Il existe une variante de ce syndrome appelé dysplasie campomélique « acampomélique » qui se distingue par l’absence d’incurvation des os longs. Quelques cas ont été décrits. Les nouveau-nés atteints de syndrome campomélique décèdent souvent rapidement après la naissance dans un tableau de détresse respiratoire, mais environ 5 à 10% des individus atteints survivent.
Apparaissent alors des complications telles qu’une cyphoscoliose, des infections respiratoires récurrentes, une perte d’audition, des difficultés d’apprentissage légères à modérées, une petite taille et une dislocation des hanches. Le traitement est symptomatique. *Auteur : Orphanet (janvier 2007).
Dysplasie osseuse létale type de La Chapelle
Dysplasie spondylo-epiphysaire tardive avec laxité ligamentaire
Embryo fœtopathie par aminopterine
Fibrochondrogenèse
La fibrochondrogenèse est une chrondrodysplasie rhizomélique néonatale, rare et létale. Onze cas ont été rapportés.
Elle se caractérise par des yeux protubérants, un étage moyen de la face et un petit nez plat avec des narines antéversées et une petite bouche avec un philtrum long. D’autres anomalies peuvent être présentes telles une fente palatine, une micrognathie et une langue bifide. Tous les segments des membres, inférieurs et supérieurs, sont très raccourcis, avec néanmoins des bras et des pieds d’une taille relativement normale. Aucune anomalie interne, à part une omphalocèle, n’a été rapportée.
La transmission de ce syndrome est probablement autosomique récessive.
Un phénomène de récurrence a été rapporté dans une famille consanguine où des personnes des deux sexes étaient atteintes, de même qu’une concordance chez des jumeaux de sexe masculin. *Auteur : Pr L. Al-Gazali (février 2005).
Maladie de Kuskokwin
Maladie des brides amniotiques
La maladie dite des brides amniotiques inclut une série de malformations dont la dénomination est liée à une hypothèse étiologique sous-jacente. Les anomalies seraient dues à une rupture de la membrane amniotique au premier trimestre de la gestation, avec pour conséquences, des adhérences entre l’amnios et le revêtement cutané du foetus, un engagement d’une partie du foetus dans la brèche, et la formation de brides à partir de l’amnios déchiré et du mésenchyme chorionique.
Ce mécanisme pourrait expliquer les anneaux de constriction autour des membres et/ou des doigts, les pseudo-syndactylies, les amputations distales des membres et aussi les constrictions du cordon ombilical. On peut également observer des déformations secondaires à la diminution des mouvements foetaux en cas de brides reliant une partie de membre et l’amnios ou en liaison avec un oligoamnios par rupture des membranes, et dans ce cas l’hypoplasie pulmonaire est fréquente.
La diminution des mouvements foetaux peut avoir d’autres conséquences : scoliose, déformations du pied, oedèmes.
Cette affection est rare, elle atteint entre 1 et 4 nouveau-nés sur 100 000.
Elle est en principe sporadique, mais des constellations intra-familiales ont été décrites, faisant évoquer une hérédité dominante. Le tableau clinique ne permet pas de différencier les formes génétiques des formes sporadiques, et la caractéristique des brides amniotiques est d’atteindre des parties de membres aléatoires : on n’observe pratiquement jamais de malformations identiques chez deux sujets atteints.
C’est l’examen du placenta et des annexes qui fait le diagnostic, lorsque des brides ou des résidus de membrane amniotique sont présents sur la face placentaire d’implantation du cordon ombilical. La maladie des brides amniotiques doit être distinguée du syndrome d’Adams Oliver, dominant autosomique, qui associe des amputations de membres de gravité diverse à une aplasie circonscrite ostéo-cutanée du cuir chevelu. Bien que le débat sur la pathogénie de la maladie des brides amniotiques persiste dans la littérature, la plupart des auteurs admettent qu’elle doit aussi être différenciée de celle de l’association « limb body wall complex », qui associe à des amputations sévères des membres des défauts de fermeture majeurs de la paroi thoraco-abdominale, des exencéphalies ou encéphalocèles et des fentes faciales différentes des classiques fentes labiales ou labio-palatines.
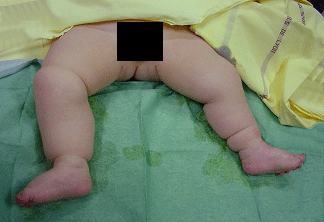
Maladie neuromusculaires
Dystrophie myotonique
Multiple core disease
Maladie Charcot-Marie-Tooth type IA
Myasthénie grave
Nanisme diastrophique
La dysplasie diastrophique ou nanisme diastrophique est une affection rare responsable d’un nanisme (taille adulte 120cm +/- 10cm) et de limitations articulaires. La maladie touche les deux sexes. A la naissance, le nouveau-né a des pieds bots, des membres courts, une déformation du poignet, souvent une fente palatine et une hypoplasie mandibulaire.
Dans les premiers mois, des kystes du pavillon de l’oreille apparaissent. La croissance est lente. La scoliose est fréquente et doit être traitée par corset. Les déformations articulaires sont importantes, soit limitées (flessum), soit au contraire hyperlaxes et font l’objet d’interventions chirurgicales parfois répétées. La transmission de la maladie est récessive autosomique. Il s’agit de mutations homozygotes du gène DTDST, transporteur de sulfates, particulièrement exprimé dans le cartilage et localisé sur le chromosome 5. Ce gène est également impliqué dans les affections létales telles que l’achondrogenèse de type Ib et l’atélostéogenèse de type II appelée aussi maladie de la Chapelle. Ces affections sont incompatibles avec la vie et repérées in utero par échographie. En revanche d’autres mutations de ce gène peuvent être aussi responsables d’une dysplasie épiphysaire modérée peu invalidante. Le diagnostic est radiologique : os courts massifs, métaphyses élargies, aspect ovoïde du premier métacarpien, points carpiens visibles à la naissance. Une subluxation des vertèbres cervicales peut être présente et justifie la surveillance de la stabilité rachidienne. L’expression de la maladie est variable avec des formes très sévères et d’autres très modérées dont le diagnostic peut être très tardif. Mais au sein d’une même famille on constate la même sévérité.
Nanisme dyssegmentaire
Nanisme pseudo-diastrophique
Ostéogenèse(s) imparfaite(s)
Groupe d’affections héréditaires de gravité variable, caractérisées par une fragilité osseuse, à l’origine de déformations du squelette et de fractures.
Ces maladies sont secondaires à un problème de fabrication (en quantité ou qualité) d’une protéine : le collagène
Ceci est à l’origine d’anomalies des os (ostéoporose) et d’anomalies des autres tissus.
Pterigia multiples
Pterigia multiples létaux
La forme létale du syndrome des pterygium multiples (LMPS) fait partie des tableaux malformatifs comprenant des pterygium (ponts cutanés limitant la mobilité des articulations) et dont l’hétérogénéité est apparue dans les années 80, lors des autopsies pratiquées de façon quasi-systématique en cas de décès d’un foetus ou d’un nouveau-né malformé.
Ce syndrome est à différencier en particulier de celui de Bartsocas Papas, syndrome létal dans lequel les pterygium ne sont localisés qu’au niveau poplité, ainsi que des formes non létales du syndrome des pterygium multiples et du syndrome des pterygium poplités, et enfin de la séquence dite « akinésie foetale » ou syndrome de Pena-Shokeir type I.
La forme létale du syndrome des pterygium multiples associe à un retard de croissance intra-utérin des pterygium marqués de multiples localisations (entre le menton et le sternum, au niveau cervical, axillaire, huméro-cubital, crural, poplité, et au niveau des chevilles) et des contractures de ces articulations donnant un tableau d’arthrogrypose sévère. Un oedème est toujours présent, allant du simple oedème sous-cutané à l’anasarque avec hygroma kystique, hydramnios et hypoplasie pulmonaire. Les anomalies faciales incluent un hypertélorisme avec épicanthus et racine du nez aplatie, une fente palatine avec micrognathie et microstomie, des fentes palpébrales obliques en bas et en dehors et des oreilles bas implantées et mal ourlées. De multiples autres anomalies ont été décrites : petit thorax, ectopie testiculaire, anomalies des dermatoglyphes, et de façon plus inconstante hémangiome du front, malrotation intestinale, hernie diaphragmatique, uropathie obstructive, microcéphalie ou atrophie ponto-cérébelleuse.
Deux cas d’hyperthermie maligne avant le décès ont également été rapportés chez un frère et une soeur.
La radiographie foetale permet de définir deux formes distinctes du syndrome : l’une avec fusion spinale et l’autre avec fusions entre des os longs. Dans la forme avec fusion spinale, les membres sont très courts et larges. La radiographie montre des fusions des apophyses épineuses des vertèbres, mais également des synostoses huméro-radiale ou radio-cubitale. Seulement deux tels cas ont été décrits, et le mode de transmission n’est pas connu. Dans la forme avec fusion des os longs, les membres sont moins courts, fins, avec peu de masse musculaire, et la fusion est visible entre les cartilages d’os longs (fémur/tibia) de forme normale.
Au total, 47 foetus ou nouveau-nés présentant un LMPS ont été identifiés dans 28 familles. Parmi eux, 28 étaient de sexe masculin et 19 féminin. Dans 14 de ces 27 familles, il n’y avait que des garçons atteints, et dans 5 d’entre elles plusieurs garçons étaient atteints. Dans une autre famille 4 garçons atteints sont nés de parents non consanguins.
Ces observations ont fait évoquer par certains auteurs une hérédité récessive liée à l’X, ce qui remet en question l’idée précédemment admise d’une récessivité autosomique. En cas d’existence d’un cas index, le diagnostic prénatal est fait à l’échographie du 2ème trimestre sur l’hygroma colli et l’anasarque, mais s’il s’agit d’un premier cas, le diagnostic peut n’être fait que plus tard.
Le caractère toujours létal à court terme du syndrome mène à proposer alors l’interruption de grossesse. *Auteur : Orphanet (juillet 2005).
Syndrome 13q-
Syndrome 4p-
Syndrome de Carraro
Syndrome de Freeman-Sheldon
Le syndrome de Freeman-Sheldon (SFS), aussi connu comme « syndrome de la face du siffleur », est un syndrome congénital rare caractérisé par une dysmorphie combinant des anomalies osseuses et des contractures articulaires avec un faciès typique. Le SFS fait partie du groupe nosologique des arthrogryphoses. Les trois anomalies caractéristiques sont une microstomie avec des lèvres en U, une camptodactylie avec une déviation cubitale de la main et un pied bot varus équin. Le phénotype du SFS inclut également une scoliose, un sillon mentonien en H, des plis naso-labiaux profonds et un blépharophimosis. Une dysphagie, un retard de croissance et des complications pulmonaires potentiellement mortelles dues à des altérations structurelles de l’oropharynx et des voies aériennes supérieures sont fréquents. Jusqu’en 1990 seulement 65 cas avaient été rapportés dans la littérature. Les deux sexes sont également touchés. La plupart des cas de SFS sont sporadiques sans aucune notion d’histoire familiale, bien qu’il existe des cas décrits compatibles avec une hérédité autosomique dominante ou récessive. L’étiologie demeure inconnue. L’échographie (révélant des anomalies des membres et de la bouche) peut aider le diagnostic prénatal. Compte tenu de sa variabilité clinique et de sa rareté, il n’y a pas de traitement standardisé pour le SFS. La correction chirurgicale de la microsmie est importante dans un but aussi bien esthétique que fonctionnel. Une chirurgie plastique et reconstructrice multiple est souvent nécessaire. *Auteur : Orphanet (mars 2005).
Syndrome de German
Le syndrome de German est caractérisé par l’association d’hypotonie et d’hypokinésie, d’une arthrogrypose et d’un lymphoedème.
Au total cinq cas ont été décrits, deux étant frère et soeur. Trois des quatre familles ayant un enfant atteint étaient juives ashkénazes. Un garçon est décédé à deux ans d’un cor pulmonale, une fille était mort-née.
La transmission de ce syndrome est probablement récessive autosomique. *Auteur : Orphanet (novembre 2005).
Syndrome de Gordon
Le syndrome de Gordon est un syndrome génétique extrêmement rare caractérisé par l’association d’une camptodactylie (fixation permanente de plusieurs doigts en position fléchie), de pied bot (incurvation anormale du pied vers l’intérieur) et dans 25% des cas, d’une fente palatine.
L’intelligence est normale, mais dans certains cas d’autres anomalies peuvent s’additionner, comme une scoliose ou une cryptorchidie. Le nombre et la sévérité des symptômes varient d’un patient à l’autre.
Le syndrome de Gordon semble être une maladie à transmission dominante, autosomique ou lié à l’X, avec une pénétrance incomplète (plus faible chez les femmes que chez les sujets de sexe masculin) et une expression variable. L’étiologie reste inconnue. Auteur : Orphanet (février 2005).
Syndrome de Larsen
Le syndrome de Larsen est une affection génétique rare caractérisée par la luxation congénitale de nombreuses articulations, une hypermobilité articulaire importante, et des traits du visage caractéristiques (arête nasale plate, hypertélorisme, et éventuellement fente palatine).
Des problèmes respiratoires dus au manque de rigidité des voies aériennes supérieures peuvent survenir.
Le syndrome de Larsen concerne environ une naissance sur 100 000. Plus de 40 enfants atteints ont été rapportés sur l’Ile de la Réunion, où l’incidence est de 1 naissance sur 1500.
Le syndrome de Larsen peut être transmis sur le mode autosomique dominant ou récessif. Des mutations du gène FLNB codant pour la filamine B (localisé en 3p14.3) sont responsables de la forme à transmission dominante et probablement des quelques cas sporadiques.
La prise en charge est adaptée à chaque cas et peut inclure un traitement orthopédique, une intervention chirurgicale en plusieurs temps, une respiration assistée, une rééducation du langage, et une kinésithérapie. *Auteur : Orphanet (mai 2006).
Syndrome de Marden-Walker
Marden et Walker ont décrit en 1966 un nourrisson de sexe féminin souffrant d’un blépharophimosis, de contractures articulaires, d’arachnodactylie et d’un retard de la croissance et du développement ; cette petite fille est décédée d’une pneumonie à l’âge de 3 mois. Depuis 1966, environ 30 cas du syndrome de Marden-Walker ont été reportés.
Les signes sont principalement présents au cours de la période néo-natale et les manifestations les plus fréquentes comprennent des contractures articulaires congénitales multiples, des caractéristiques dysmorphiques avec une expression faciale figée, un blépharophimosis, un ptosis, une micrognathie, une fente palatine ou un palais rehaussé, des oreilles bas implantées, une arachnodactylie, une masse musculaire accrue.
L’évolution de la maladie est toujours caractérisée par un retard de croissance et un retard psychomoteur. Initialement décrite comme un syndrome, cette affection correspondrait plutôt à une expression phénotypique de différentes maladies hétérogènes et appartient au groupe II des arthrogryposes.
Le mécanisme pathologique sous-jacent n’a pas été clairement établi. Le diagnostic est uniquement basé sur les critères cliniques.
Le traitement est uniquement symptomatique, avec une gestion multidisciplinaire (kinésithérapie, psychomotricité, orthophonie). *Auteur : Dr D. Héron (novembre 2003)*
Syndrome de Noonan
Le syndrome de Noonan est caractérisé par une petite taille, une dysmorphie faciale caractéristique et des anomalies cardiaques congénitales.
La prévalence à la naissance varie entre 1/1 000 et 1/2 500 naissances vivantes.
Les principaux traits du visage associés au syndrome de Noonan sont un hypertélorisme avec des fentes palpébrales antimongoloïdes, un ptosis, des oreilles bas implantées en rotation postérieure avec un hélix épais. Les anomalies cardiovasculaires les plus souvent associées sont une sténose pulmonaire et une cardiomyopathie hypertrophique. Les patients présentent aussi un cou palmé court, une déformation de la cage thoracique, une déficience intellectuelle légère, une cryptorchidie, des difficultés d’alimentation durant la petite enfance, une tendance aux saignements et des dysplasies lymphatiques.
Syndrome de Pena-Shokeir I
La séquence d’akinésie foetale est évoquée devant une arthrogrypose avec ankylose de toutes les articulations des membres, camptodactylie, le tout associé à une hypoplasie pulmonaire et à une dysmorphie faciale.
Il s’agit d’une séquence dont le premier élément est l’immobilité foetale, quelle qu’en soit la cause. L’absence de mouvements de déglutition induit un hydramnios, et la diminution des mouvements du diaphragme et des muscles intercostaux est à l’origine de l’hypoplasie pulmonaire, en règle sévère. La diminution des mouvements foetaux induit également une arthrogrypose multiple et une relative brièveté du cordon. Les mains sont en coup de vent cubital, les pieds sont bots et souvent en piolet, les dermatoglyphes sont peu abondants et les plis de flexion des doigts et des paumes sont effacés. Les 1er et 2ème doigts de la main chevauchent les doigts 3, 4 et 5. La face est inexpressive, rigide, avec hypertélorisme, télécanthus, oreilles petites, mal ourlées et en rotation postérieure, petite bouche avec palais ogival et micrognathie. Plus rarement, on peut voir une fente palatine et/ou une cardiopathie.
La prématurité est fréquente, et même en cas de naissance à terme les nouveau-nés ont un retard de croissance intra-utérin, une cryptorchidie, un cou court en apparence et lorsqu’ils survivent, un syndrome du grêle court avec malabsorption. L’ensemble des signes doit conduire à un diagnostic différentiel avec la trisomie 18 par l’obtention d’un caryotype.
Le syndrome de Pena-Shokeir est rare, décrit dans une centaine de cas dans la littérature. Environ 30% des enfants sont mort-nés, et une majorité des enfants nés vivants décèdent après quelques semaines de leur hypoplasie pulmonaire.
Les causes et mécanismes physiopathologiques de cette séquence malformative sont hétérogènes. Dans environ 50% des cas, il s’agit d’un syndrome récessif autosomique (consanguinité parentale), dans d’autres cas on retrouve une myasthénie maternelle ; l’expérimentation animale suggère que le curare administré à la mère pourrait aussi avoir cette conséquence, et toutes les autres causes plausibles d’immobilité (myogène, neurogène, ischémique/anoxique) doivent être recherchées sur des biopsies.
Cette hétérogénéité rend le conseil génétique difficile, et conduit à indiquer, devant un cas isolé sans étiologie évidente un risque de récurrence compris entre 0,01 et 25%.
Le diagnostic prénatal, après la naissance d’un cas index, repose sur une échographie morphologique à faire vers la 18ème semaine d’aménorrhée, un hydramnios avec diminution/disparition des mouvements foetaux, un oedème du scalp et une hypoplasie pulmonaire doivent être recherchés. *Auteur : Orphanet (juillet 2005).
Syndrome de Roberts
Le syndrome de Roberts a été décrit pour la première fois par Roberts en 1919 et se manifeste par une dysmorphie faciale, et des anomalies réductionnelles des membres, voisines de la phocomélie. Environ 100 cas ont été décrits.
Les éléments dysmorphiques sont un visage arrondi, des yeux globuleux, une bouche ronde et une fente labiopalatine. L’atteinte des membres se traduit par un raccourcissement des os longs qui intéresse préférentiellement le segment mésomélique mais aussi parfois le segment proximal, la main ou le pied. Le membre supérieur est le plus souvent touché et certaines pièces osseuses peuvent être absentes. Des incurvations des os et une flexion irréductible des articulations sont possibles.
L’observation de récurrence dans une même fratrie est en faveur d’un mode d’hérédité autosomique récessif mais le gène à l’origine de la maladie n’est pas localisé ou identifié à ce jour.
Ce gène est probablement responsable d’anomalies mitotiques. En effet, l’étude cytogénétique des patients porteurs d’un syndrome de Roberts montre des anomalies chromosomiques impliquant les régions hétérochromatiques autour des centromères et des organisateurs nucléolaires. L’hétérochromatine des bras longs du chromosome Y est aussi souvent anormalement séparée durant la métaphase.
Ces anomalies suggèrent une répulsion ou une absence d’attraction des régions chromatidiennes conduisant à une séparation prématurée durant la prophase et la métaphase. Le syndrome pseudothalidomide ou syndrome SC est probablement une forme modérée de syndrome de Roberts. Enfin, le syndrome TAR (Thrombopenia-Absent radius syndrome) s’en distingue par les manifestations hématologiques et la rareté des fentes mais la distinction en 2 entités différentes est discutée. *Auteur : Dr V. Cormier-Daire (septembre 2002).
Syndrome de Zellweger
Le syndrome cérébro-hépato-rénal de Zellweger est caractérisé par l’association de :
1) un début anténatal avec anomalies morphogénétiques comportant notamment une dysmorphie faciale très caractéristique et des anomalies de migration neuronale ;
2) une hypotonie néonatale sévère avec aréflexie, épilepsie et nystagmus sans aucune acquisition psychomotrice ;
3) des troubles neuro-sensoriels (cataracte, choriorétinite, atrophie optique, surdité neuro-sensorielle) ;
4) une petite taille avec des ponctuations épiphysaires, cryptorchidie chez les garçons ou hypertrophie clitoridienne chez les filles ;
5) des anomalies hépato-rénales comportant des kystes hépatiques et rénaux, une fibrose ou une cirrhose et une absence de peroxysomes morphologiquement repérables ;
6) le décès survient généralement avant l’âge de 6 mois ;
7) toutes les fonctions biologiques peroxysomales sont déficitaires (accumulation d’acides gras à très longue chaîne, de précurseurs des acides biliaires, acide pipécolique, acide phytanique, déficit de synthèse des plasmalogènes et déficit généralisé des protéines de la bêta-oxydation). *Auteur : Orphanet (septembre 2002).
Syndrome du pouce adductus
Syndrome Fémoro-facial
Syndrome Hydro-létal
Syndrome Oro-facio-digital IV
Triploïdie
Trisomie 7
Trisomie 8
Anémie de Fanconi
Maladie autosomique récessive associée à une instabilité chromosomique, l’anémie de Fanconi (AF) est marquée par une hétérogénéité phénotypique qui inclut une insuffisance médullaire, un syndrome malformatif variable, une propension à développer des leucémies aiguës myéloïdes (LAM) et une hypersensibilité cellulaire aux agents pontant l’ADN.
Cette dernière caractéristique permet d’étudier les mécanismes à la base de la maladie et sert aussi au diagnostic clinique.
L’AF a été trouvée dans tous les groupes ethniques, son incidence est estimée à 1/350 000 naissances, elle est cliniquement caractérisée par une pancytopénie, une anémie aplasique progressive, diverses malformations congénitales (malformations du squelette, hyperpigmentation, anomalies urogénitales, rénales et cardiaques) et surtout une prédisposition élevée aux LAM.
Les troubles hématologiques dus à un dysfonctionnement de la moelle osseuse n’apparaissent en moyenne que vers l’âge de 7 ans, ils peuvent exceptionnellement se révéler très tôt dès la naissance ou plus rarement très tard à l’âge de 40 ans.
La greffe de moelle osseuse ou de sang de cordon ombilical reste à ce jour le principal traitement relativement efficace de la défaillance hématologique typique de l’AF.
La thérapie cellulaire à l’aide de cellules souches isolées ainsi que la thérapie génique font l’objet de recherches qui n’ont pas encore abouti de façon concluante.
Il existe à ce jour 8 groupes de complémentation et 6 gènes ont été clonés FANCA, FANCC, FANCD2, FANCE, FANCF et FANCG sur différents chromosomes. Il manque encore une vision unificatrice précise des événements biochimiques en cause dans l’AF. La fonction des différents gènes FANC reste inconnue.
La similitude globale des phénotypes cliniques et surtout cellulaires laisse penser que les protéines participent à un même réseau métabolique majeur. *Auteur : Dr E. Moustacchi (octobre 2003).
Anophtalmie avec anomalie des membres
Arachnodactylie avec contracture
Arthro-osteo-onychodysplasie
Brachydactylie type C
La brachydactylie de type C (BDC) est une malformation congénitale très rare caractérisée par une brachymésophalangie de l’index, du majeur et du petit doigt, une hyperphalangie de l’index et du majeur, et par un rétrécissement du premier métacarpe.
Peu de cas ont été rapportés dans la littérature.
L’annulaire est en général le doigt le plus long. Des métacarpes petits et une symphalangie sont parfois décrits.
Des mutations hétérozygotes au niveau du gène>, nommé growth/differentiation factor-5 gene (GDF5), codant pour la protéine cartilage-derived morphogenetic protein 1 ont été rapportées.
Plusieurs études suggèrent une hérédité autosomique dominante. *Auteur : Orphanet (mai 2008).
Chondrodysplasie ponctuée
Dysplasie chondro-ectodermique
Le syndrome d’Ellis-van Creveld (EVC) est une dysplasie chondro-ectodermique caractérisé par des côtes courtes, une polydactylie, un retard de croissance, des anomalies ectodermiques et des malformations cardiaques.
C’est une affection rare, avec environ 150 cas rapportés dans le monde. Sa prévalence exacte est inconnue, mais le syndrome semble plus fréquent dans la communauté Amish.
Après la naissance, les manifestations majeures sont une petite taille, des côtes courtes, une polydactylie, et une dysplasie des ongles et des dents. Des malformations cardiaques, et particulièrement des anomalies des cloisons interauriculaires, surviennent dans 60% des cas. Le développement psychomoteur est normal.
Cette affection rare se transmet sur le mode autosomique récessif avec une expression variable. Des mutations des gènes EVC1 et EVC2, situés sur le chromosome 4p16 et de sens de lecture opposé, sont responsables du syndrome. Les anomalies prénatales (pouvant être détectées à l’échographie) incluent un thorax étroit, un raccourcissement des os longs, une hexadactylie et des malformations cardiaques.
Le syndrome d’EVC appartient au groupe des syndromes côtes courtes-polydactylie qui doivent être évoqués lors du diagnostic prénatal (surtout le syndrome de Verma-Naumoff de type III ; voir ce terme).
Après la naissance, le diagnostic différentiel comprend la dystrophie de Jeune, le syndrome de McKusick-Kaufman et le syndrome de Weyers (voir ces termes).
La prise en charge du syndrome d’EVC est pluridisciplinaire. En période néonatale, la prise en charge est principalement symptomatique, incluant le traitement des problèmes cardiaques et de la détresse respiratoire due à l’étroitesse de la cage thoracique. Un suivi orthopédique est nécessaire en raison des déformations osseuses. Les manifestations orales de la maladie doivent faire l’objet de soins dentaires.
Le pronostic dépend des difficultés respiratoires rencontrées dans les premiers mois de vie et des éventuelles malformations cardiaques. Il est difficile d’estimer la taille définitive des personnes atteintes. *Auteurs : Drs G. Baujat et M. Le Merrer (juin 2007) Traduction Orphanet.
En savoir plus sur la Dysplasie chondro-ectodermique ou Syndrome d’Ellis-van Creveld
Dysplasie spondylo-epiphysaire congénitale
Embryo-foetopathie alcoolique
Embryo-foetopathie par les hydantoïnes
Gangliosidose GM1
La gangliosidose à GM1 est une maladie neurodégénérative se caractérisant par une accumulation du ganglioside GM1.
On distingue 3 types de gangliosidose à GM1.
Le type infantile (type I) débute dans le premier trimestre de la vie par une encéphalopathie évolutive avec amaurose. Il existe d’emblée une hépatosplénomégalie et une infiltration cutanéo-muqueuse entraînant un visage aux traits grossiers et des déformations du squelette dont une cyphoscoliose. Un retard voire un arrêt du développement (suivi d’une détérioration neurologique progressive) survient généralement dans les six premiers mois de vie. Dans 50% des cas une tache rouge cerise au fond d’oeil est retrouvée. Les oligosaccharides urinaires sont abondamment sécrétés.
Le type juvénile (type II) survient dans la deuxième enfance (1-5 ans). Il se traduit par une ataxie locomotrice et évolue vers un état de décérébration avec crises d’épilepsie. L’atteinte viscérale est discrète. Dans la forme de l’adulte (type III ou gangliosidose à GM1 chronique), le début d’apparition est variable, parfois juvénile, mais le diagnostic n’est souvent porté qu’à l’âge adulte. Les signes cliniques sont ceux d’une maladie de Parkinson juvénile, d’une dégénérescence spino-cérébelleuse atypique ou d’une dystonie. Il n’y a pas d’atteinte viscérale ni de tache rouge cerise. Le déficit intellectuel peut être initialement absent ou léger mais progresse avec le temps. La gangliosidose à GM1 est due à un déficit en bêta-galactosidase (bêta-gal) lysosomale.
Elle se transmet sur le mode récessif autosomique.
Le gène responsable est localisé sur le chromosome 3 (3p21.33). Une douzaine de mutations ont été identifiées ; elles empêchent la phosphorylation du précurseur de la bêta-galactosidase, qui est sécrété au lieu d’être acheminé dans les lysosomes. Le déficit en bêta-gal et l’accumulation de GM1 semblent induire indirectement l’activation d’une voie apoptotique neuronale. Le diagnostic peut être confirmé par la biopsie de peau et par la mesure de l’activité de la bêta-galactosidase qui est très diminuée dans les leucocytes ou les fibroblastes cultivés.
Le dépistage des hétérozygotes et le diagnostic prénatal sont possibles.
Le pronostic varie en fonction de l’âge de début de la gangliosidose. L’espérance de vie ne dépasse pas deux ans dans la forme infantile, et rarement plus de vingt ans dans la forme juvénile.
Pour la forme adulte, le phénotype est variable, mais l’apparition progressive de séquelles neurologiques aboutit généralement à une réduction de l’espérance de vie.
Un traitement qui vise à inhiber la synthèse des gangliosides (Miglustat) est à l’étude pour les formes lentement évolutives. *Auteurs : Prs N. Baumann et J-C. Turpin (avril 2006).
Hemi-hypertrophie congénitale
Lépréchaunisme
Le lépréchaunisme est une maladie congénitale caractérisée par un retard de croissance intra-utérin et surtout post-natal majeur.
Cette maladie est très rare avec moins de 1 cas par million de naissances.
Le retard staturo-pondéral s’associe cliniquement à un faciès dysmorphique caractéristique (rappelant les gnomes du folklore irlandais ou « leprechaun »), une atrophie du tissu adipeux sous-cutané (lipoatrophie) et une hypotrophie musculaire.
Chez la petite fille, des signes de virilisation sont souvent observés. Biologiquement, des épisodes d’hypo- et d’hyperglycémie sont observés avec constamment un hyperinsulinisme majeur traduisant un état d’insulino-résistance extrême.
La transmission de cette maladie est de type autosomique récessif. Au niveau génétique, la présence de mutations du récepteur de l’insuline est nécessaire au diagnostic soit homozygote soit hétérozygote composite.
Le diagnostic positif nécessite de retrouver une mutation dans chaque allèle de ce gène. Le pronostic est réservé avec des difficultés de croissance et une espérance de vie qui dépasse rarement quelques mois. Un traitement avec l’IGF-1 (Insulin-like growth factor I) recombinant pourrait éventuellement être envisagé.
Une association de IGF-BP3 (insulin-like growth factor binding protein 3) a amélioré la durée de vie dans un cas. *Auteur : Pr J. Capeau (février 2005).
Maladies des synostoses multiples
Osteodysplastie
Polyomélite
La poliomyélite est une maladie infectieuse virale provoquée par l’un des trois sérotypes du poliovirus humain, virus qui fait partie de la famille des entérovirus.
Elle débute la plupart du temps par une paralysie aiguë flasque.
Elle se manifeste principalement chez l’enfant âgé de moins de 5 ans. La transmission est essentiellement inter-humaine, notamment via la voie oro-fécale.
Généralement, l’infection est limitée à la voie gastro-intestinale et au rhinopharynx, et est souvent asymptomatique. Le système nerveux central, notamment la moelle épinière, peut être affecté, générant rapidement une paralysie progressive.
Les neurones moteurs sont essentiellement touchés. Une encéphalite peut également survenir.
Le virus se multiplie dans le système nerveux et touche particulièrement les neurones moteurs de la corne antérieure de la moelle épinière (« polio » signifiant « gris »).
La poliomyélite doit être distinguée des autres états paralytiques par l’identification du virus dans les selles.
La prévention est l’unique traitement contre la poliomyélite paralytique.
Deux vaccins contre la poliomyélite sont disponibles : le vaccin inactivé injectable et le vaccin vivant atténué en prise orale.
En France, la vaccination obligatoire chez l’enfant depuis les années 60 a fait disparaître cette maladie redoutable. Les progrès effectués dans le cadre de l’éradication mondiale de la poliomyélite, depuis leurs débuts en 1988, ont été remarquables.
En 1988, 125 pays étaient endémiques pour la poliomyélite et on estimait à 1000 le nombre d’enfants paralysés chaque jour par le poliovirus sauvage.
Fin 2003, on ne recensait plus que six pays endémiques pour la poliomyélite (Afghanistan, Egypte, Inde, Niger, Nigeria et Pakistan) et le poliovirus paralysait moins de 3 enfants par jour. L’interruption de la transmission par l’homme du poliovirus sauvage dans le monde entier a été fixé comme l’objectif à atteindre pour fin 2004.
Ptegyrum poplite
Syndrome Cérébro-costo-mandibullaire
Syndrome d’Elhers-Danlos
Syndrome de Catel-Manzke
Le syndrome de Catel-Manzke est caractérisé par une association malformative comprenant une micrognathie avec glossoptose et fente palatine (séquence de Pierre Robin), et une anomalie des 2 index (présence d’un petit os inséré dans l’articulation métacarpo-phalangienne, entraînant une déviation radiale des index).
Il a été décrit dans environ 25 cas.
Outre ces deux signes principaux, plusieurs types de malformations associées ont été décrits, principalement cardiopathies et retards de croissance.
D’autres anomalies ont été décrites, chez l’un ou l’autre des cas rapportés : colobome de l’iris, hypertélorisme, fentes palpébrales étroites, oreille bas implantée et en rotation postérieure, brachydactylie, clinodactylie des 5ème doigts, pectus excavatum, genoux luxables, hallux courts ou scoliose.
Une ventriculomégalie cérébrale a été décrite une fois. Le développement des enfants a été décrit dans 10 cas, et il existait un retard mental chez 2 d’entre eux, modéré chez l’un et sévère chez l’autre. Les autres enfants étaient dans les limites de la normale. L’étiologie génétique proposée aujourd’hui est celle d’une microdélétion, avec un syndrome dit des gènes contigus, mais cette hypothèse ne pourra être confirmée que par l’examen cytogénétique de nouveaux cas avec des techniques avancées.
Parce que les cas premiers décrits n’étaient que des garçons, une hérédité de type récessive liée à l’X a été proposée. Plusieurs filles atteintes ont ensuite été rapportées, y compris 2 soeurs, excluant le mode de transmission d’abord envisagé, d’autant plus qu’un cas a été décrit de transmission père-fils. *Auteur : Orphanet (octobre 2004).
Syndrome de Fraser
Le syndrome de Fraser est une entité clinique rare dont les signes cardinaux sont la cryptophtalmie et la syndactylie.
En tout environ 150 patients atteints ont été décrits dans la littérature.
La cryptophtalmie est présente dans 93% des cas. Il s’agit d’une absence de fentes palpébrales avec fissures, microphtalmie sévère ou anophtalmie, et absence ou malformation des canaux lacrymaux. La syndactylie est présente dans 54% des cas.
Des anomalies urinaires (reins absents ou multikystiques) et beaucoup d’autres malformations ont été décrites dans ce syndrome: malformations de l’oreille moyenne ou de l’oreille externe, palais ogival, clivage le long de la paroi inter-narinaire ou de la langue, hypertélorisme, sténose laryngée, diastème de la symphyse pubienne, déplacement de l’ombilic ou des mamelons, mésentère commun. Chez les filles, on a décrit des fusions labiales, hypertrophies clitoridiennes, utérus bicornes et malformations des trompes de Fallope ; chez les garçons les anomalies génitales décrites incluent des ectopies testiculaires et des micropénis avec hypospadias. La plupart des patients n’ont pas de déficience intellectuelle, mais sont polyhandicapés.
Vingt-cinq pour cent des enfants sont morts-nés, et 20% décèdent avant l’âge de 1 an, à cause des anomalies rénales ou laryngées. Si ces anomalies ne sont pas présentes, l’espérance de vie est pratiquement normale.
Environ 15% des cas sont nés de couples consanguins, le mode de transmission du syndrome étant autosomique récessif.
Le diagnostic échographique du syndrome de Fraser est possible à 18 semaines de grossesse.
Il doit être évoqué lorsqu’au moins deux des signes suivants sont détectés : microphtalmie, syndactylie, gros poumons hyperéchogènes, hydramnios.
Le gène du syndrome de Fraser a été localisé en 4q21. Au moins 5 mutations ont été identifiées à l’intérieur de ce gène FRAS1, qui code probablement pour une protéine de la matrice extracellulaire. *Auteur : Orphanet (mars 2006).
Syndrome de Goldenhar
Le syndrome de Goldenhar ou dysplasie oculo-auriculo-vertébrale (OAV) est caractérisé par une hypoplasie faciale asymétrique associée à des anomalies oculaires (dermoïde ou lipodermoïde épibulbaire, microphtalmie, colobome de la paupière supérieure…), une microtie, des appendices ou sinus pré-auriculaires et des malformations vertébrales (fusion des cervicales, platybasie, puzzle vertébral…).
L’atteinte faciale est généralement unilatérale, mais peut être bilatérale avec une expression plus sévère d’un côté. Une microtie avec ou sans appendice prétragien est considérée par certains comme une expression minimale de dysplasie OAV.
D’autres malformations sont rapportées dans 50% des cas : anomalies cardiaques, cérébrales, rénales, gastro-intestinales.
Un retard mental n’est présent que dans 10% des cas. Il existe un risque d’apnées obstructives dans le sommeil.
La prévalence de la dysplasie OAV est estimée entre 1/5 600 et 1/20 000 naissances. La plupart des cas sont sporadiques et le risque de récurrence empirique est faible (2-3%). Dans les cas familiaux, le mode de transmission est compatible avec une hérédité autosomique dominante. La dysplasie OAV apparaît comme un défaut de champ de développement embryonnaire complexe.
L’hétérogénéité génétique est certaine. *Auteur : Pr D. Lacombe (janvier 2004).
Syndrome de Marshall-Weaver
Syndrome de Moebius
Le syndrome de Moebius correspond à une paralysie congénitale des muscles des yeux et du visage.
Environ 300 cas ont été décrits dans la littérature.
Le premier symptôme est une difficulté à téter. Les nouveau-nés peuvent aussi baver de manière excessive et présenter un strabisme. Ensuite, c’est l’absence d’expression du visage (et de sourire notamment), et l’absence de clignements et de mouvements latéraux des yeux qui dominent le tableau clinique.
D’autres anomalies sont parfois associées, comme une déformation de la langue (et donc des difficultés d’élocution), de la mâchoire ou encore des malformations des membres chez un tiers des patients. Elles peuvent inclure un pied bot, des doigts manquants ou palmés et une anomalie de Poland, entre autres.
La plupart des enfants souffrent d’une hypotonie musculaire, plus particulièrement dans la partie supérieure du corps, entraînant un retard d’apprentissage de la marche. Un léger déficit intellectuel se manifeste chez environ 10% des patients.
Le syndrome de Moebius est dû à une anomalie de développement du 7ème nerf crânien (facial) dans tous les cas, et dans 75% des cas du 6ème (abducens). D’autres nerfs crâniens peuvent être affectés plus occasionnellement (notamment les 3ème, 4ème, 5ème, 9ème, 10ème et 12ème).
La plupart des cas sont sporadiques sans histoire familiale particulière. La maladie n’est pas progressive, et la prise en charge est surtout symptomatique.
La nutrition de ces enfants peut nécessiter un biberon particulier ou une alimentation par sonde. Le strabisme, les anomalies des membres ou de la mâchoire peuvent être corrigés chirurgicalement. Les patients peuvent aussi bénéficier d’une rééducation physique et orthophonique pour améliorer leurs capacités motrices et leur coordination et leur permettre de mieux maîtriser le langage et l’alimentation. Une greffe de muscle peut procurer une certaine mobilité au visage et donner la capacité de sourire. *Auteur : Orphanet (août 2006).
Syndrome de Robin
Syndrome de Smith-Lemli-Opitz
Le syndrome de Smith Lemli Opitz (SLO) associe une dysmorphie faciale (microcéphalie, microrétrognatisme, implantation basse des oreilles, anomalies du philtrum et du palais), des anomalies cérébrales (troubles de la myélinisation, agénésie du corps calleux, holoprosencéphalie) et un retard statural.
Les difficultés nutritionnelles et le retard psychomoteur sont très variables. Un défaut de masculinisation des organes génitaux externes (voire une ambiguïté sexuelle) et une anomalie des extrémités à type de syndactylie 2-3 sont des signes de grande valeur diagnostique à la naissance et même avant lors de l’examen échographique.
La prévalence est estimée entre 1/30 000 et 1/10 000 depuis qu’un test chimique permet le diagnostic positif chez l’enfant en période prénatale. Les sujets homozygotes présentent une diminution du cholestérol et une accumulation de stérols aberrants, dérivés des précurseurs dans la voie de biosynthèse (7-déhydrocholestérol , 8-déhydrocholestérol, nortriénol, dérivés oxydés…). La maladie est due à des mutations récessives du gène de l’enzyme 7-déhydrocholestérol réductase porté par le chromosome 11 en 11q13.
La supplémentation diététique enrichie en cholestérol (1 à 2 jaunes d’oeuf), en acides biliaires (acide ursodésoxycholique) et l’inhibition en amont de la synthèse endogène de stérols aberrants (statines sont conseillées avec des bénéfices variables sur la croissance, l’humeur et la photosensibilité de l’enfant.
Le SLO est transmis sur un mode récessif avec un risque de récurrence de 25% en cas de nouvelle grossesse, ce qui impose la surveillance écho-morphologique précise des grossesses (retard de croissance intra-utérin, anomalies de la face et des extrémités).
Le diagnostic anténatal par étude biochimique du liquide amniotique est très fiable et peut se faire dès 11 semaines par choriocentèse.
Le génotype permet la confirmation ou l’exclusion du diagnostic chez l’enfant et chez les éventuels porteurs de mutations (les sujets hétérozygotes ne présentent pas la maladie).
Le séquençage génétique confirme la fréquence de 45% environ de la mutation du site d’épissage de l’intron 8 dans le gène de l’enzyme 7-dehydrocholestérol réductase pour la population des patients étudiés en France. L’analyse suggère une corrélation entre la position de la mutation sur l’exon codant pour la fixation du co-substrat de l’enzyme (NADPH), sa nature (non-sens, délétion, stop) et la sévérité du phénotype associé.
Les recherches actuelles portent sur la synthèse résiduelle de cholestérol et l’intensité du phénotype. *Auteur : Dr C. Wolf (octobre 2004).
Trisomie 13
Dernière MàJ : 26/01/2019